药群论坛
标题: 2022液质联用经验汇总,必须收藏! [打印本页]
作者: xiaoxiao 时间: 2022-5-13 03:48 PM
标题: 2022液质联用经验汇总,必须收藏!
2022液质联用经验汇总,必须收藏!
一、液质使用经验与禁忌
1、酸性物质适合做负离子检测,所以流动相偏碱性较合适,促使其解离,碱性物质适合做正离子检测,流动相中适当的加入酸,促使其形成正离子,流动相中适当加一些醋酸钠(或者醋酸铵),可形成加钠的正离子或者加铵的正离子。
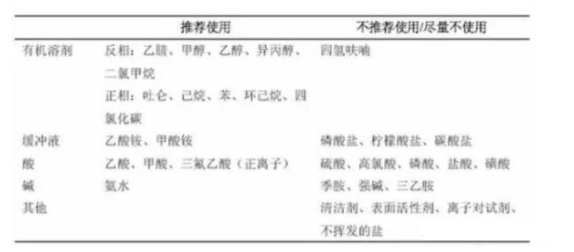
液质分析中推荐使用的流动相和添加剂
2、糖苷类的物质在做FAB和esi(+)时,[M+Na]峰往往比[M+H]峰要强,此为经验,原因只是推测可能和天然产物的提取过程有关;盐类化合物如盐酸盐、硫酸盐在质谱中酸的部分一般不会出现;二羧酸盐(esi负离子模式)除了分子离子峰外,会出现连续掉44的两个峰,为失去羧酸根的离子,这三个峰非常特征,但是会受锥孔电压的影响,调低电压谱图会更漂亮。
3、胺类物质做esi质谱时要注意进样量要少,因为很容易离子化,不易冲洗干净,会影响后面样品的测定。像三乙胺在液质联用时不能用于调节流动相pH值。若不慎引入三乙胺,在正离子检测时总会出现很强的102峰(三乙胺的[M+H])。
4、质谱用水一般用娃哈哈纯净水之类的就很好;质谱用甲醇和乙腈,换用了很多品牌,发现Merck的还是稍微好一些;Finnigan用的氮气不一定要用到液氮瓶,用普通的钢瓶气就可以了,可能还省钱些;建议大家买一个好一点的手电筒和一个放大镜,手电筒用来看源里面,放大镜看你割的毛细管平整。
5、质谱的基线其实跟液相的紫外检测器和荧光检测器一样,基线高的原因不外乎内部和外部的原因。
1)你选择的流动相在质谱的响应比较高,比如水相比较多的时候,噪音比较大些;还有如果盐含量比较大的时候,噪音更大些。
2)检测器的灵敏度越高的时候,噪音应该越高。如果质谱的污染比较严重时,基线肯定比较高。比如离子阱检测器,用得久了,阱中的离子就会增多,一方面降低了质谱的灵敏度,另一方面增加了基线噪音。
3)质谱的基线很多时候还跟你选择的离子宽度有关。比如你作选择离子扫描的时候,基线就低些。你作选择反应扫描的时候,离子宽度不要选得太宽,太宽噪音就高些。
4)多级质谱一般做二级或3J质谱,基线噪音就低很多。
6、质谱维护经验交流:做样前-检查氮气,流动相,质谱仪的真空度,毛细管温度…
1)最好不用直接进样(容易污染离子源);
2)做联用时最好分流(a可以使用常规柱,b缩短分析时间,c 延长质量分析器寿命);
3)最好使用在线切换阀,降前每个样品的前后1-2分钟的流动相切入废液(避免样品中的盐进入质谱,做Sequence时可以把平衡柱子的流动相切入废液);
4)开始联用前,直接运行质谱数分钟,可以先将温度(毛细管温度和离子源温度(APCI))加热到预设定值(如果是APCI源还可以避免将烧掉heater,太贵了,最好别烧);
5)待机时将切换阀置于waste,避免刚开液相时将流动相打入离子源;
6)关机前毛细管的温度先降下来,稳定一段时间后再关闭电源,避免风扇停止转动后毛细管外围的热量向里扩散,容易引起内部线路及电子元器件老化加速;
7)每天清理毛细管口外部,擦洗干净,每次停机时注意清洗Skimmer,用无尘擦拭纸,kimberly那种;
8)如果用的是钢瓶而且天天做样的话,将两个钢瓶并联,当然,一月不做一次的话就算了;
9)做定量时注意离子源喷针的具体位置,否则标准曲线就不能用了;
10)不要不经过柱子分离进行定量分析,结果不可靠(竞争性抑制目标分子离子化);
11)如果是负离子检测的话,可以相流动相中加入少量异丙醇;
12)不要使用不挥发性盐,如果使用挥发性盐,但浓度不要超过20mmol/l;
13)需要使用酸的情况下可以用甲酸,乙酸,三氟乙酸可以用,但能用甲酸或乙酸时就别用TFA;
7、理论上液质联用禁止使用任何不挥发性的缓冲盐,如果需要尽量使用诸如乙酸氨等挥发性盐,浓度不要超过20mmol/l。
对于不挥发性的缓冲盐,如果你的仪器有吹扫捕集的话也可使用,但一定要小心。万不得已也不要用,首先有不挥发盐是得不到好的离子流的,其次盐留在质谱中很难除掉,除非停机清洗,不然一直会影响其他样品的分析。
可以找质谱友好的条件来做液质联机,例如色谱条件为20mM磷酸盐的水/乙腈流动相,做液质联机的时候就可以用醋酸铵代替,然后用醋酸调节pH值与磷酸盐的一致即可。
除了难挥发的盐,三乙胺、表面活性剂、还有高浓度(>0.5%)的TFA,都对质谱不好,液质联用的流动相中应该避免。
二、质量定量分析经验
1、要用目标离子的碎片定量,特征性强,排除干扰;
2、在定量分析的方法设置上,尽可能提高扫描速率,提高准确率和重复性(可以通过a减小扫描质量数的范围来提高目标峰的扫描次数,或 将一个样品全部分析时间断分成n个segments,对目标离子单独设置扫描模式);
3、一定要通过色谱柱分离后定量分析,避免竞争性离子的存在影响目标离子的离子化效率;如果目标分子未与竞争性分子完全分开,则在离子化过程中导致目标分子的离子化效率降低,导致样品分子的定量结果偏低,当然标准浓度的样品也要用相同的方法分析;
4、如果样品都是纯品的话可以不经过色谱柱直接进样分析,包括做标准曲线的样品(虽然不建议直接进样分析);
5、如果用的离子源的喷针位置是可移的话,一定要记住做标准曲线时其位置,否则其位置移动后在相同的条件下进入质谱的离子流量会发生改变,标准曲线就不能使用了!对于调用的质谱方法不要改动shealth gas and aux gas 的流速,否则会影响进入质谱的样品量;
6、所建立的标准曲线一个月后如果想重复使用,则用QC样品检验一下该标准曲线;
7、对于已经建立好的分析方法在扫描范围、流动相的组成、梯度或流速等方面不要作任何改动,否则,标准曲线要重作。扫描范围改变目标峰的扫描次数、流动相组成改变离子化效率,流速改变色谱峰的保留时间和峰宽;
8、离子阱的强项在于多级-定性,四级杆的强项在于定量;
9、对于热稳定性不好的样品可以通过提高气速,降低毛细管温度的方法保证定量分析的重复性;一旦方法固定后不要轻易改动。
三、解析图谱
解析未知样的质谱图,大致按以下程序进行。
(一)解析分子离子区
(1) 标出各峰的质荷比数,尤其注意高质荷比区的峰。
(2) 识别分子离子峰。首先在高质荷比区假定分子离子峰,判断该假定分子离子峰与相邻碎片离子峰关系是否合理,然后判断其是否符合氮律。若二者均相符,可认为是分子离子峰。
(3) 分析同位素峰簇的相对强度比及峰与峰间的Dm值,判断化合物是否含有Cl、Br、S、Si等元素及F、P、I等无同位素的元素。
(4) 推导分子式,计算不饱和度。由高分辨质谱仪测得的精确分子量或由同位素峰簇的相对强度计算分子式。若二者均难以实现时,则由分子离子峰丢失的碎片及主要碎片离子推导,或与其它方法配合。
(5) 由分子离子峰的相对强度了解分子结构的信息。分子离子峰的相对强度由分子的结构所决定,结构稳定性大,相对强度就大。对于分子量约200的化合物,若分子离子峰为基峰或强蜂,谱图中碎片离子较少、表明该化合物是高稳定性分子,可能为芳烃或稠环化合物。
例如:萘分子离子峰m/z 128为基峰,蒽醌分子离子峰m/z 208也是基峰。
分子离子峰弱或不出现,化合物可能为多支链烃类、醇类、酸类等。
(二)解析碎片离子
(1) 由特征离子峰及丢失的中性碎片了解可能的结构信息。
若质谱图中出现系列CnH2n+1峰,则化合物可能含长链烷基。若出现或部分出现m/z 77,66,65,51,40,39等弱的碎片离子蜂,表明化合物含有苯基。若m/z 91或105为基峰或强峰,表明化合物含有苄基或苯甲酰基。若质谱图中基峰或强峰出现在质荷比的中部,而其它碎片离子峰少,则化合物可能由两部分结构较稳定,其间由容易断裂的弱键相连。
(2) 综合分析以上得到的全部信息,结合分子式及不饱和度,提出化合物的可能结构。
(3) 分析所推导的可能结构的裂解机理,看其是否与质谱图相符,确定其结构,并进一步解释质谱,或与标准谱图比较,或与其它谱(1H NMR、13C NMR、IR)配合,确证结构。
--- THE END ---
免责声明:本文系转载分享,版权归原作者所有。如涉及版权,请联系删除!
| 欢迎光临 药群论坛 (http://yaoqun.net/) |
Powered by Discuz! X3.2 |