药群论坛
标题: 关联审评一年回顾:企业为何跟进缓慢? [打印本页]
作者: 静悄悄 时间: 2018-12-24 10:10 AM
标题: 关联审评一年回顾:企业为何跟进缓慢?
来源:医药经济报
在国家药监局官网上,可搜索到“关联审评”的两个关键文件,分别是2016年8月10日发布的<总局关于药包材药用辅料与药品关联审评审批有关事项的公告(2016年第134号)>,以及2017年11月30日发布的<总局关于调整原料药、药用辅料和药包材审评审批事项的公告(2017年第146号)>。
可以看出,一开始关联审评并没有将原料药包含在内,而是在药包材、药用辅料和药品联合审评试行逐渐稳定后,才将原料药纳入其中,期间观察了一年多时间。这既跟上了国际药品审评先进政策的步伐,也显示出了国家药监部门对一项新政策推行的谨慎认真态度。
从“4+7”试点集采结果看,原料药制剂一体化企业未来在竞争中会有很大优势。不过从审批数据看,2017年年底才正式公布的关联审评决定,实施一年来却进展缓慢,原因何在?
A 原料药政策过渡影响大
药审中心官网中原料药登记数据库中现共有2614条记录,但其中已激活并批准完成与上市制剂联合审评审批的只有156条,占比仅为6.2%,大部分原料药依然处于未与制剂完成关联的状态,甚至还有19条原料药已经失效。
过渡期影响涉及各方
这个结果看起来似乎不太尽如人意,但考虑到药品审评所需时限和政策过渡的影响,也是可以接受的。
2017年12月1日起,国家局调整和实施药品注册受理工作新的职能范围,由国家局审评审批、备案的注册申请,均改为国家局直接受理。包括新药临床试验申请、新药生产(含新药**)申请、仿制药申请,国家局审批的补充申请等。由省级食品药品监督管理部门审批、备案的药品注册申请仍由省级食品药品监督管理部门受理。
国家药监局这项职能调整在关联审评政策发布后的第二天就开始实施了,体现了其对药品审评新政的重视。不过,大量药品注册资料直接由国家局受理,无疑加大了国家局的工作量。再加上处于政策过渡阶段,药企和药审部门都需要一定的时间去学习和适应新的药审工作程序,这些都不同程度地影响到药审工作效率。
企业方面,尽管对于关联审评,前期已有大量风声放出,但在没有正式文件发布之前,也没有几家药企敢于贸然提前布局。
一来国家政策当时尚未明确,大部分原料药企业并没有专门的下游制剂生产商,政策公布后再去寻求制剂厂商合作,既费时又费力。
二来许多制药企业原料药的再注册批件尚未到有效期,按照规定还是具有法律效应的。因此,这类企业暂不急于找制剂厂商进行合作。
当然,也有制剂企业不愿轻易改变原料药来源的因素。毕竟重大变更需要重新进行生产工艺验证、现场和抽样检查,甚至是加速和长期稳定性实验,一系列工作会让制剂停产至少半年时间。这些显然是制剂厂商无法接受的,除非能够带来远超投入的收益。
不过,随着未来政策的不断实施推进,完成与制剂关联审评的原料药登记数量将明显上升。
进一步完善的规定
政策中规定,药品注册申请人在境内提出的注册分类2.2、2.3、2.4、3、4、5类药品制剂申请所使用的原料药均要与相应制剂关联审评。至于1类和2.1类的创新药和革新药,原本就有规定需要在药品申请时同时提交原料药加制剂申请。
虽然原料药由原先的“审批制”改为“先登记再与制剂一并审评”,但资料主要内容并无变化,基本信息、生产信息、特性鉴定、质量控制、对照品、药包材、稳定性等缺一不可。此外,还需按年度提交产品质量管理报告,在产品发生变更时应及时在登记平台中变更相关信息,并在实施变更前主动告知使用其产品的药品制剂申请人。
因此,整体来说,此次政策的改进,将药品的审评要求进一步严格完善化了。
B 药用辅料部分辅料可免登记
药用辅料在药审中心共有1209条记录,其中已激活并批准完成与上市制剂联合审评审批的只有15条,占比仅为1.2%,数量和占比量都远远落后于原料药。不过值得庆幸的是,暂时还没有失效的药用辅料产品。
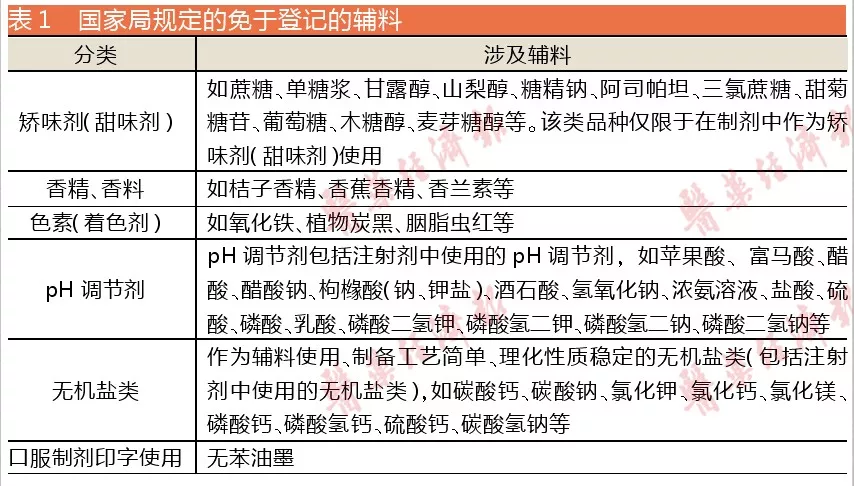
完成与制剂关联审评的药用辅料数量少的原因,与原料药基本一致;至于整体登记数量依然比原料药少,则是因为有部分药用辅料产品制备工艺简单、长期使用且质量标准稳定等,国家局给予免于登记的特权(具体见表1)。
对于这些免于登记的药用辅料,现行版<中国药典>已收载的,应符合现行版<中国药典>要求;现行版<中国药典>未收载的,应符合国家食品标准或现行版美国药典/国家处方集、欧洲药典、日本药典、英国药典标准要求;其他辅料,应符合药用要求。
药用辅料登记资料主要内容包括企业基本信息、辅料基本信息、生产信息、特性鉴定、质量控制、批检验报告、稳定性研究、药理毒理研究等,要求参照国家局通告2016年第155号中的药用辅料申报资料。同样,也要按年度提交产品质量管理报告,以及在产品发生变更时及时在登记平台中变更相关信息,并在实施变更前主动告知使用其产品的药品制剂申请人。
C 药包材相容性研究成难点
药包材数据库目前共有2602条数据,其中只有3条已完成关联审评,数量占比是三者中最少的,仅有0.1%。
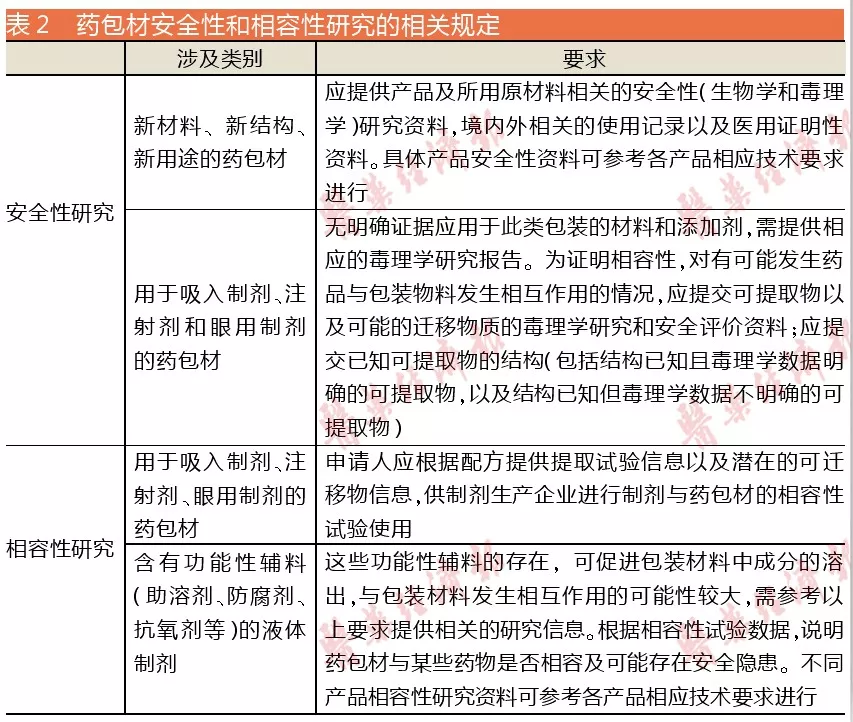
除了与原料药和药用辅料的共性原因外,还有一点重要原因或难点是药包材安全性和相容性研究的要求较之前有所提高,许多药包材企业并没有相关研究经验,只能向第三方机构寻求帮助。但目前相关案例并不多,大家也都还在探索阶段。
2016年第155号文对药包材申报资料要求(试行)中,对药包材安全性和相容性研究的要求见表2。
可以看出,对于药包材安全性和相容性研究这方面,国家还没有太明确的要求,只是规定了适用的范围。搜索国家药监局官网,与药包材相容性有关的现行指导原则也只有两条,分别是2012年9月发布的<化学药品注射剂与塑料包装材料相容性研究技术指导原则>,以及2015年7月发布的<化学药品注射剂与药用玻璃包装容器相容性研究技术指导原则>,都是试行文件。而具体如何操作实施,还是需要药包材和制剂企业按照药品特性自行研究发掘。
目前来说,药品与包装材料的相容性研究,应在药品研发初期或包装材料选择时就开始进行,并贯穿于药品研发的整个过程。
首先,应对包装组件所用材料以及添加剂等进行分析,然后通过初步的稳定性试验、加速试验和长期稳定性试验考察包装材料对药品稳定性的影响,并通过药物与包装材料的相容性研究考察包装材料中成分迁移进入药品的程度、包装材料对制剂中活性成分与功能性辅料的吸附程度,确认包装材料可保证药品质量稳定,并与药品相容性良好。
上市后,如需变更包装,则应评估该变更对药品质量可能产生的影响,并根据影响程度设计相关的试验进行研究,特别是应进行变更后包装材料与药品的相容性研究,证明这种变更不足以对药品质量以及包装材料功能性产生不可接受的变化,即不会导致安全性风险。
对于塑料材料,相容性研究内容主要包括提取实验、相互作用研究(迁移试验和吸附试验)和安全性研究三个方面。对于玻璃材料,相容性研究内容主要包括模拟试验和相互作用研究,针对不同的待测项目要选择专属性强、准确、精密、灵敏、适宜的经过分析方法验证的方法。
结语<<<
关联审评政策已经施行一年多时间,目前来看进展缓慢。而随着相关政策和指导原则的不断完善,以及先前原辅料和药包材“政策福利”逐渐到期,再加上一致性评价工作和MAH制度对高质量药用原辅料和药包材需求的不断提高,这一系列因素将促使更多的原料药、药用辅料和药包材企业积极与药用制剂厂商的合作,并不断提高自身产品的质量。
作者: LNPH 时间: 2018-12-24 05:58 PM
本帖最后由 LNPH 于 2018-12-24 06:01 PM 编辑
另外7+4之后,感觉企业的积极性进一步下滑
作者: jack757 时间: 2018-12-25 10:51 AM
好文章,谢谢楼主精辟分析。
作者: 279660321 时间: 2020-1-13 03:46 PM
谢谢楼主分享
作者: haixin86 时间: 2021-3-23 04:47 PM
好文章,谢谢楼主分析分享。
作者: mengmeng11 时间: 2023-9-21 09:58 AM
谢谢楼主分享
| 欢迎光临 药群论坛 (http://yaoqun.net/) |
Powered by Discuz! X3.2 |