日前,瑞士制药公司Helsinn集团和位于圣地亚哥的MEI Pharma公司联合宣布,欧洲药品管理局(EMA)授予两个公司联合开发的在研药物pracinostat孤儿药资格。
Pracinostat目前在临床3期试验中与azacitidine构成组合疗法,用于治疗无法接受诱导化疗的成年急性骨髓性白血病 (acute myeloid leukemia, AML) 患者。EMA的孤儿药资格是基于治疗AML患者疗法的稀缺和pracinostat在临床2期试验中的积极表现。
AML是成年人中最常见的急性白血病,而且随着世界人口的老龄化它的发病率预计会继续增长。美国癌症学会 (American Cancer Society) 估计2017年美国有21380名新的AML患者,有10590名患者因此去世。目前美国FDA还未批准任何疗法治疗那些无法接受强化诱导化疗的AML患者。
▲Pracinostat的分子结构式(图片来源:Selleck Chemicals)
Pracinostat是最初由MEI Pharma公司开发的口服组蛋白去乙酰化酶 (histone deacetylase, HDAC) 抑制剂。HDACs能够通过化学修饰DNA和与之结合的染色体蛋白来调控基因表达,这些表观遗传学调节因子的异常活性在癌症中起到很重要的作用。美国FDA在2016年8月授予pracinostat和azacitidine组合疗法突破性疗法认定。Azacitidine是一种化疗药物,它可以作为低甲级化剂来治疗AML。MEI Pharma 公司在2016年8月与Helsinn集团达成合作协议共同开发pracinostat。
在已经完成的开放标签,多中心,单臂临床2期试验中,pracinostat和azacitidine被用于治疗50名65岁以上,不适于接受诱导化疗的AML患者。试验结果表明,接受这一组合疗法的患者的中位总生存期为19.1个月,一年生存率为62%,完全缓解率为42%。这一表现显著好于azacitidine单一疗法在类似患者中的疗效(中位总生存期为10.2个月,完全缓解率为19.5%)。
Helsinn集团的CEO兼副主席Riccardo Braglia先生说:“我们很高兴EMA能够授予pracinostat孤儿药资格。这一决定激励我们继续投入大量资源来加快我们的临床试验项目,早日帮助像AML患者一样与罕见难治疾病作斗争的患者。”
Helsinn集团和MEI Pharma公司已经启动检验pracinostat和azacitidine组合疗法的关键性临床3期试验,去年7月,第一名患者已经开始接受治疗。
原标题:总生存期几乎翻倍,白血病新药获孤儿药资格
参考资料:
[1] Helsinn Group and MEI Pharma Announce that Pracinostat has Received Orphan Drug Designation from the European Medicines Agency for the Treatment of Acute Myeloid Leukemia (AML)
[2] MEI Pharma 官网
----------------------------------------------------------------------
5、镰状红细胞病新药voxelotor获FDA突破性疗法认定
Global Blood Therapeutics(GBT)近日宣布,美国FDA为镰状红细胞病(SCD)新药voxelotor(以前称为GBT440)颁发了突破性疗法认定(BTD)。Voxelotor作为正在开发的SCD缓解疗法,已获得了欧洲药品管理局(EMA)的优先药物(PRIME)资格。Voxelotor是首个获得突破性疗法认定的治疗SCD在研新药。
SCD是由血红蛋白β链中的基因突变引起的终身遗传性血液疾病,会形成异常的镰状血红蛋白(HbS)。在脱氧状态下,HbS具有聚合或结合在一起的倾向,在红细胞内形成刚性棒状结构。聚合的棒状结构使红细胞变成镰刀状和不灵活,可导致毛细血管和小血管的堵塞。从儿童期开始,SCD患者由于血液无法顺畅流向器官,受到不可预知和反复发作的严重疼痛或危机,常导致心理和身体残疾。阻断的血流,加上溶血性贫血(红细胞的破坏),最终可能导致多器官损伤和早期死亡。
Voxelotor正在开发成为每日一次的口服疗法,治疗SCD患者。Voxelotor通过增加血红蛋白对氧的亲和力达到疗效。由于氧合镰状血红蛋白不会聚合,voxelotor可防止红细胞发生镰状改变。Voxelotor有着恢复正常血红蛋白功能和改善氧气输送的潜力,GBT认为voxelotor可能改变SCD的疾病进程。FDA已经认识到对SCD新疗法的迫切需求,为voxelotor颁发了快速通道资格,孤儿药和罕见小儿疾病资格。
▲GBT的研发管线(图片来源:GBT官网)
此项BTD的决定基于voxelotor提交的几项临床研究数据,其中包括:3期研究HOPE A部分的初步有效性和安全性数据(GBT440-031),1/2期的成人开放标签扩展研究(GBT440- 001/024),在6至17岁儿童中进行的2期研究HOPE-KIDS 1(GBT440-007),以及在患有严重SCD成年人中的同情使用。
其中,在最近一项公开的临床研究GBT440-007发现,儿童和青少年在治疗16周后,血红蛋白水平增加,溶血症状的临床指标得到改善。结果与先前在成人中观察到的结果一致。具体而言,16周时55%的患者(11位患者中的6位)达到了>1g/dL的血红蛋白缓解,中位血红蛋白变化为1.1g/dL。根据总症状评分(TSS)评估,16周时12名患者中有10名每日症状得到改善。这一发现表明,患者报告的结果(PRO)测量对治疗效果敏感,并支持其正在进行的3期HOPE(血红蛋白氧亲和调节抑制HbS聚合作用)研究中使用。药物在青少年中耐受,没有与药物相关的严重不良事件或中断使用。
“FDA授予voxelotor第一个治疗镰状红细胞病的突破性疗法认定的决定,反映了FDA对这种在研新药有效性和安全性数据的认可,以及对SCD患者改善目前疗法需求的认知,”GBT公司总裁兼首席执行官Ted W. Love先生说:“随着我们正在加快voxelotor的发展,这项认定是GBT的另一个重要里程碑。”
我们期待这款突破性疗法在临床试验中能够取得更多的突出表现,为患有这种影响广泛疾病的患者缓解疾病进程,提高生活质量。
原标题:FDA颁发首款镰状红细胞病突破性疗法认定
参考资料:
[1] FDA Calls Bay Area Global Blood Therapeutics' Sickle Cell Drug a Breakthrough
[2] GBT Receives FDA Breakthrough Therapy Designation for Voxelotor for Treatment of Sickle Cell Disease (SCD)
[3] GBT Announces New Phase 2a Data at ASH for Voxelotor in Adolescents with Sickle Cell Disease (SCD)
--------------------------------------------------------
6、默沙东新型HIV药物Doravirine在美进入正式审查
美国制药巨头默沙东(Merck & Co)近日宣布,美国食品和药物管理局(FDA)已受理新型HIV药物doravirine(MK-1439,DOR)的2份新药申请(NDA)。DOR是一种实验性非核苷类逆转录酶抑制剂(NNRTI),目前正在多个临床研究中进行开发,用于HIV-1成人感染者的治疗。这2份NDA中,其中一份申请将DOR作为一种每日一次的单一片剂,联合其他抗逆转录病毒药物,为患者提供一种量身定制的完整治疗方案;另一份申请将DOR与拉米夫定(3TC)和替诺福韦酯(TDF)组成的固定剂量组合单一片剂(DOR/3TC/TDF),作为一种每日一次的完整治疗方案。FDA已指定这2份NDA的处方药用户收费法(PDUFA)目标日期为2018年10月23日。
默沙东研究实验室全球临床开发副总裁George Hanna博士表示,自从HIV流行以来,默沙东一直致力于研究和满足HIV群体的需求。DOR是该公司开发出的一种新的HIV药物,将为患者提供一种有意义的新治疗选择,解决该领域存在的未满足医疗需求。
DOR两份NDA的提交,是基于2项正在开展的III期临床研究(DRIVE-FORWARD,DRIVE-AHEAD)的数据。这2个研究分别评估了DOR分别联合2种背景NRTI组合的完整治疗方案以及DOR/3TC/TDF固定剂量组合单一片剂完整治疗方案的疗效和安全性。除了这些研究之外,默沙东正在开展另一项III期临床研究DRIVE-SHIFT,在接受其他抗逆转录病毒方案实现病毒学抑制的HIV-1成人感染者中,评估转向DOR/3TC/TDF方案的疗效和安全性。
DRIVE-FORWARD:DOR单片联合背景NRTI疗效强劲,血脂参数更优
DRIVE-FORWARD(NCT02275780)是一项多中心、双盲、随机、非劣效性研究,在769例既往未接受抗逆转录病毒药物治疗(初治)HIV-1成人感染者中开展。入组该研究的患者在治疗前的HIV-1 RNA≥1000拷贝/毫升,研究中,这些患者以1:1的比例随机接受DOR(100mg剂量,每日一次),或利托那韦(ritonavir)增效的达芦那韦(darunavir)(DRV+r,800mg+100mg,每日一次),各组同时接受研究者所选择的2种常用NRTI背景方案(TDF/FTC或ABC/3TC,每日一次),治疗直至96周。该研究的主要终点是治疗第48周时实现HIV RNA<50拷贝/毫升的患者比例,预先定义的非劣效性边界值为10%。次要终点包括DOR和DRV+r对空腹血脂的影响、CD4+T细胞计数从基线的变化,耐受性。
研究结果显示:(1)769例受试者中有766例接受了研究药物(每个治疗组383例)并纳入了疗效和安全性分析(平均年龄35.2岁,84%男性,73%白人,87%接受TDF/FTC)。在治疗第48周,DOR治疗组实现HIV-1 RNA<50拷贝/毫升的患者比例为83.8%(n=321/383),DRV+r治疗组为79.9%(n=306/383,差异=3.9%,95%CI[-1.6,9.4]),数据达到了非劣效性标准。(2)在基线HIV-1 RNA>100000拷贝/毫升的亚组中,治疗第48周时,DOR治疗组和DRV+r治疗组实现HIV-1 RNA<50拷贝/毫升的患者比例分别为81.0%(n=64/79)和76.4%(n=55/72)。(3)主要安全性终点方面,不良事件发生率(总体、严重、药物相关、治疗停药)在DOR治疗组和DRV+r治疗组相似,最常见的药物相关不良事件(>5%)为腹泻(5.5% vs 12.8%)、恶心(6.5% vs 7.6%)和头痛(6.0% vs 2.6%)。(4)次要终点方面,DOR治疗组空腹LDL-C和non-HDL-C水平相对基线降低(分别为:-4.5mg/dL和-5.3mg/dL),DRV+r治疗组空腹LDL-C和非-HDL-C水平相对基线均表现升高(分别为:+9.9mg/dL,+13.8mg/dL),数据具有统计学显着差异(空腹LDL-C:差异=-14.6mg/dL,95%CI[-18.2,-11.1],p<0.0001;空腹non-HDL-C:差异=-19.3,95%CI[-23.3,-15.4],p<0.0001)。
该研究证实:在初治HIV-1成人群体中,与2种常用背景NRTI方案联合用药时,在治疗第48周,DOR表现出了强劲的疗效,与DRV+r相比具有非劣效性,并与基线HIV-1 RNA水平无关。安全性方面,DOR的安全性和耐受性良好,与DRV+r相比具有更优越的血脂参数属性。
DRIVE-AHEAD:DOR/3TC/TDF复方单片疗效强劲,神经精神安全性更好,血脂参数更优
DRIVE-AHEAD(NCT02403674)是一项多中心、随机、双盲、非劣效性研究,在734例既往未接受抗逆转录病毒药物治疗(初治)HIV-1成人感染者中开展,评估了每日一次固定剂量组合单一片剂DOR/3TC/TDF(100mg/300mg/300mg)相对于每日一次固定剂量组合单一片剂EFV/FTC/TDF(600mg/200mg/300mg)的疗效和安全性。入组该研究的患者在治疗前的HIV-1 RNA≥1000拷贝/毫升,研究中,这些患者以1:1的比例随机接受DOR/3TC/TDF或EFV/FTC/TDF治疗直至96周,随机化分组通过HIV-1 RNA水平(≤/>100000拷贝/毫升)和乙肝/丙肝共感染(是/否)进行分级。研究的主要疗效终点是治疗第48周时HIV RNA<50拷贝/毫升的患者比例,预先定义的非劣效性边界值为10%。主要安全终点是治疗48周期间发生下述神经精神不良事件的患者比例:头晕、睡眠障碍和紊乱、感觉改变。次要终点包括:DOR/3TC/TDF和EFV/FTC/TDF对空腹血脂的影响、CD4+T细胞计数从基线的变化,耐受性。
研究结果如下所示:(1)734例受试者中有728例接受了研究药物(每个治疗组364例)并纳入了分析(平均年龄33岁,85%男性,48%白人)。在治疗第48周,DOR/3TC/TDF治疗组实现HIV-1 RNA<50拷贝/毫升的患者比例为84.3%,FEV/FTC/TDF治疗组为80.8%(差异=3.5%,95%CI[-2.0,9.0])。(2)主要安全性终点方面,DOR/3TC/TDF治疗组头晕、睡眠障碍和紊乱、感觉改变发生率比EFV/FTC/TDF治疗组低(分别为:8.8% vs 37.1%[p<0.001],12.1% vs 25.5%[p<0.001],4.4% vs 8.2%[p=0.033])。(3)次要终点方面,DOR/3TC/TDF治疗组空腹LDL-C和non-HDL-C水平相对基线降低(分别为:-1.6mg/dL和-3.0mg/dL),EFV/FTC/TDF治疗组空腹LDL-C和非-HDL-C水平相对基线均表现升高(分别为:+0.7mg/dL,+13.3mg/dL),数据具有统计学显着差异(空腹LDL-C:差异=-10.0mg/dL,95%CI[-13.5,-6.5],p<0.0001;空腹non-HDL-C:差异=-17.0,95%CI[-20.9,-13.2],p<0.0001)。
该研究证实:在初治HIV-1成人群体中,在治疗第48周,DOR/3TC/TDF单一片剂方案与EFV/FTC/TDF单一片剂方案在疗效方面具有非劣效性和相似性,并与基线HIV-1 RNA水平无关。DOR/3TC/TDF方案的安全性和耐受性良好,神经精神事件明显少于EFV/FTC/TDF方案,同时具有更好的血脂参数。
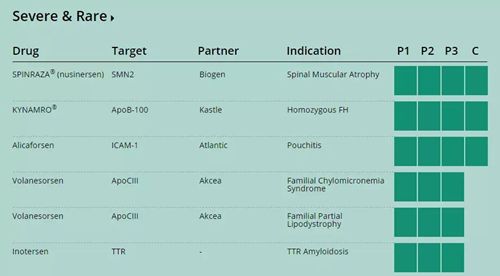
7、Ionis反义核苷酸新药获FDA优先审评资格
Ionis Pharmaceuticals今天宣布,FDA为inotersen的新药申请(NDA) 颁发了优先审评资格。Inotersen是一种用于治疗遗传性TTR淀粉样变性(hATTR)患者的在研新药。强生:制造一场危机,对投资者来说或许是好事
强生的首席执行官Alex Gorsky表示,强生应该制造一场危机,比如担心亚马逊将来会扰乱强生的业务。这样或许将帮助公司跑赢整个市场。
Gorsky关于制造危机的言论,主要是针对最近几年医疗保健领域发展的问题。他认为这是一个“非常值得你思考科学技术的时机”。
在20世纪90年代,生物制药行业因为开发对患者没有明显帮助的“me-too drugs(仿制药)”而受到了很大的抨击。但现在不同了,免疫疗法的兴起,使包括丙型肝炎和艾滋病在内的疾病得到了史无前例的治疗。
另外,强生在机器人手术这个领域也表现得非常积极。
强生与谷歌母公司Alphabet合作组建了Verb Surgical公司,致力于将机器学习、仪器、先进的可视化和数据分析结合起来,以创建一个新的机器人手术系统。Verb Surgical公司已经做出了原型,但还未推出市场。
行业变化的步伐可能在未来几年内迅速增加。这就是为什么他要求强生“制造危机”。Gorsky还特别提到亚马逊,因为越来越多的人猜测这家电子商务巨头计划进军零售药店业务和药品利益管理业务。
至于如何保持这样的危机感,Gorsky认为,首先强生需要在内部进行创新,并与外部组织合作。收购瑞士制药商Actelion便是强生如何在合作伙伴关系中发挥创造力的一个例子。
最后,Gorsky说,即使在“危机”之中,优秀的运营策略也至关重要。他认为,强生需要像“经营拖拉机工厂”那样思考。
美敦力:强调价值医疗,回应并购战略质疑
“我们发现,优化治疗是我们在价值医疗方面取得的最大成功,因为技术和技术所创造的价值是与临床结果直接相关的。”美敦力的CEO Omar Ishrak告诉投资者,“而在这方面,我们做得最好的是Tyrx,尤其是在去年,对此我们感到非常高兴。”
Tyrx是一种抗菌包膜,包裹在植入式心脏电子产品中,如心脏起搏器或除颤器,并洗脱药物以防止植入此类装置后发生感染。随着时间的推移,网状装置被完全吸收到身体中而不留下任何痕迹。
2014年,美敦力公司以1.60亿美元收购了新泽西州Tyrx公司,从而获得了该产品。
在一家大型医疗保健机构中,使用该设备一年能减少与感染有关的成本640万美元。“这是一种成功的价值医疗模式,我们正处于实施的初期,以后还将继续。”Ishrak说。
Ishrak暗示Tyrx的成功主要与医疗保健的根本驱动力有关:为期望的临床结果付费。他说:“市场迟早会有不确定性,这是一个基本的事实。在我看来,为临床结果付费,并创造附加值,这一定是正确的。”
收购Tyrx之后,Medtronic又于2015年6月宣布收购Covidien。当时,很多人对美敦力的并购战略提出了质疑。对此,Ishrak宣称他想要“遵守收购规则”,并呼吁美敦力应该保持观望,不要在购买Covidien之后为成长阶段的公司付钱。
Ishrak表示,自Covidien以后,美敦力已经花费了37亿美元在收购上,而且收购的几乎都是技术型的企业。所以,对美敦力而言,需要考虑的是如何开发一项有潜力的技术,并将其产业化。
雅培:2017完成两大并购,2018又将迎来两大里程碑事件
雅培全球执行副总裁Brian Yoor表示,进入2018年,雅培公司也迎来了两大里程碑事件。
第一,雅培成立130周年。在过去的这段时间里,雅培为医疗保健做出了伟大的贡献,并创造了巨大的股东价值。2017年,吉普林格(Kiplinger)把雅培评为有史以来最好的30只股票之一。
第二,雅培与AbbVie(艾伯维)分离5周年。这一战略创造了一个新的雅培。在宣布分离的前一天,公司的市值是820亿美元。现在,雅培和艾伯维的市值总和接近2600亿美元,增长了近220%。
在过去的一年里,雅培采取了两个最重要的战略行动,2017年1月收购圣犹达,秋季有收购了Alere(美艾利尔)。圣犹达和美艾利尔的加入强化了雅培在医疗保健关键领域的领导地位和影响力。
Alere的快速诊断技术加入到了雅培现有的POCT业务,使雅培坐上全球POCT第一的宝座。
圣犹达与雅培的血管业务相结合,使雅培成为心血管设备市场的领导者,同时还将雅培带入一个新市场,神经调节治疗慢性疼痛和运动障碍。
Brian Yoor表示,未来雅培将加大研发投入,并增加一部分投资,以巩固公司在全球市场的地位。
安进(Amgen):关于安进,投资者需要知道的5件事
2017年,生物技术的两种顶级药物Enbrel和Neulasta销售下滑,但Amgen有效控制了成本,最终收入基本持平。在J.P摩根大会上,首席执行官 Bob Bradway 就未来生物科技的未来,分享了5个观点。
1、预计2018年每股收益将实现两位数的增长。公司预计今年将削减成本约15亿美元,以提高收益。安进的股票回购也可能造成巨大的变化。
2、安进有五个“引人注目的长期增长动力”。安进公司确定了5个产品,其中骨质疏松症药物Prolia是目前公司发展最快的重磅炸弹。
3、老药不断产生强劲的现金流。尽管自身免疫性疾病药物Enbrel和Neulasta的销售额下滑,但Bradway强调,这两种药物和其他老药物对Amgen仍然很重要,这些药物预计会在未来多年产生强劲的现金流。
4、把资金还给股东是重中之重。Bradway强调:“生物技术的资本分配是一种先见之明,而不是事后的想法”。自2011年以来,安进已经为股东回报了340亿美元,其中有210亿美元的股票回购和130亿美元的派息。
5、安进将寻求什么样的收购。很多人猜测,安进公司将在不久的将来进行一次或多次收购。Bradway承认了公司扩大业务的意向,并指出“有可能会做一些有吸引力的业务发展”。
他表示,安进在任何交易中都会考虑四件事情。首先,该公司希望获得创新产品,以补充Amgen的六个治疗重点领域。其次,安进希望加快其国际战略。第三,生物技术要“利用(转化)技能”。最后,Bradway强调说,安进的任何交易都是为了给股东创造长期价值。
辉瑞:生产力提高,辉瑞的未来比过去更光明
辉瑞全球研发总裁Mikael Dolsten的发言可以总结为一个词:生产力。
辉瑞的生产力可以通过它产生多少重磅药物来判断。
2005年至2010年间,该公司推出了两款重磅产品:癌症药物Sutent和Prevnar 13肺炎球菌疫苗。
2011年到2016年,辉瑞推出了五款重磅炸弹产品。它们是肺炎球菌疫苗Prevnar 13、血液稀释剂Eliquis、类风湿性关节炎药物Xeljanz、治疗乳腺癌的药物Ibrance和治疗湿疹的药物Eucrisa。
其中,Prevnar 13肺炎球菌疫苗2017年的销售额约为54亿美元,它是辉瑞最畅销的产品。Ibrance是辉瑞增长最快的明星之一。
这种药物在2015年获得了FDA批准用于治疗乳腺癌,预计将成为排名第五的最畅销的癌症药物,未来五年内全球销售额将超过70亿美元。
Mikael Dolsten表示,辉瑞现在的生产力更高,因为公司一直专注于“严谨与科学”。这意味着辉瑞公司在评估风险和选择最有前途的资产方面做得更好。
在接下来的五年,辉瑞公司希望能够推出多达15个潜在的重磅药物。其中五个可能在肿瘤领域。它们包括Ibrance和Xtandi的新适应症,以及辉瑞公司与德国制药商Merck KGaA合作的Bavencio的组合。
最后,Dolsten并不认为收购会阻碍辉瑞的生产力进步。
他说:“对于一个大公司来说,始终关注增长或加速增长的机会是非常重要的。”辉瑞如果能够进行收购,就会迅速行动起来。例如,2016年6月辉瑞完成了对Anacor的收购,并于6个月后获得FDA的批准。
Illumina:发布新产品iSeq™ 100,牵手赛默飞世尔
由于CEO Francis de Souza在J.P.Morgan大会上的发言,Illumina股票出现了大反弹,股价早盘大涨了7%左右。Francis de Souza在会上带来了Illumina的5个好消息。
1、财务数据客观。Illumina2017年全年收入将比上年增长15%,达到27.5亿美元左右。
2、NovaSeq的持续动力。目前,Illumina已经向全球约150家客户送出了285个NovaSeq装置。Illumina希望NovaSeq有一个多年的采用周期。
公司预计目前使用Illumina的HiSeq X测序系统的850多名客户中,大部分将转去使用NovaSeq。随着基因测序成本的降低,将有更多的新客户购买NovaSeq。
3、发布iSeq™ 100测序系统。Illumina公司宣布推出一款名为iSeq的新型桌面系统,准确率高达99.8%,售价不到2万美元,2018年第一季度开始出货。公司估计,iSeq有超过5万个潜在客户,其中包括35,000名新一代测序人员。
4、主要新兴市场有了增长。De Souza表示,消费基因已经到了拐点,2017年有超过700万个消费者样品测序或基因分型。Illumina的消费基因客户是市场上的一些大牌公司,包括Ancestry、23andMe和Helix。
5、与赛默飞世尔达成合作。在JP摩根大会上,Illumina与赛默飞世尔达成合作。这使得Illumina能够将赛默飞世尔的Ion AmpliSeq技术出售给对Illumina新一代测序(NGS)平台进行科学研究的研究人员。
Ion AmpliSeq拥有一种先进的扩增子技术,能够高效的从微量的样本中DNA和RNA,适用于科研和体外诊断等多个研究领域。
礼来:100亿美元现金回笼,更多业务将得到发展
美国总统唐纳德•特朗普(Donald Trump)在12月份签署的法律的变化,位于印第安纳波利斯的礼来公司的全球税率将略微下调。因此,礼来公司将获得一笔巨额现金。
礼来公司在海外拥有100亿美元的资金,现金回笼会促进礼来公司更多业务的发展,礼来公司的竞争力也将进一步提高。
“接下来,我们可以投资建工厂,建实验室,而不用担心税率的问题。我们唯一需要担心的问题是人才和市场。”礼来公司CEO David Ricks说。
同时,Ricks还表示,他仍然想要建立更全面的产品线,特别是在癌症治疗方面。他说,现在很多有竞争力的公司都在开发免疫学、糖尿病和肿瘤学治疗业务。
默沙东:更多的药品回扣应该给到消费者
1、消费者应该享受更多的药品回扣。
大制药商默沙东(Merck)的首席执行官Ken Frazier表示:“制药公司支付的回扣,约为品牌药品公司的三分之一。有三分之一的人通过保险公司、PBMs(药店福利经理)和其他渠道,凭借回扣进入分销系统,而这些回扣是不会给消费者的。”
“我认为,我们真正要做的就是将一些回扣,在药房柜台上给到真正需要的人。”
2、如果亚马逊能让药物流通更有效,将赞成其进入药物分销领域。
Ken Frazier之所以会发表关于回扣的言论,很有可能是因为他被问及大型在线零售商亚马逊欲进入药物运送领域。他表示,如果亚马逊能让药物流通更有效,他将赞成其进入药物分销领域。
Celgene(赛尔基因):关于Celgene你不得不知的5件事
生物制药龙头Celgene的业绩在2017年出现了大幅下滑,股价下跌近10%。即使在大会之前,公司宣布收购私人生物技术公司Impact Biomedicines也不足以激发投资者,其股价在第二天早盘下跌超过3%。
尽管如此,Celgene 的首席执行官Mark Alles在JP摩根大会上也带来了几个好消息。
1. 为什么要收购Impact Biomedicines
近日,Celgene宣布拟以70亿美元收购Impact Biomedicines,但对此投资者们却并不买账。主要是因为Impact针对骨髓纤维化(MF)的靶向治疗产品fedratinib,市场上Incyte公司的Jakafi占主导地位。而且,fedratinib在去年临床上有一些潜在的安全问题。
但Alles表示,他承认Jakafi是市场的领导者,但fedratinib在治疗复发性骨髓纤维化方面仍有巨大的潜力。所有MF患者中约20%的人对Jakafi没有反应。
2、Otezla比你想象的要强
Celgene去年的糟糕表现,部分原因是Otezla的销售低于预期。但自身免疫性疾病药物并不会成为生物技术的一个阻力,Otezla在持续强劲增长方面表现良好。Alles指出了Otezla尚未开发的重大机遇,约有60%的诊断银屑病患者未经治疗。
3、期待ozanimod撼动多发性硬化症市场
Celgene 对ozanimod有很高的期望。该公司希望今年晚些时候,其用于治疗复发性多发性硬化症(MS)的药物能够获得FDA批准。如果ozanimod得到预期的批准,Alles认为它将动摇整个MS市场。
4、业绩增长稳健
虽然投资者对去年Celgene股票的表现感到失望,但公司在公布第四季度和2017年全年数据时表现出强劲的业绩。Alles表示,Celgene预计2017年全年营收将达到130亿美元,比上一年增长16%。Celgene预计2018年收入至少增长12%。
5、即使Revlimid失去独家经营权,未来仍然一片光明
一些投资者可能会担心,在公司丧失了强大的血癌药物Revlimid的排他性之后,Celgene会发生什么。Alles表现得很乐观,他表示Celgene未来五年将推出10个重磅药物,这可能会在药品的高峰期增加超过150亿美元的年度收入。
收购对Celgene的未来也很重要。随着公司在2015年收购Receptos,从而购买了Ozanimod。假设Celgene收购Impact的计划如期完成,fedratinib可能是通过收购获得的另一个重磅炸弹。
FN-1501是复星医药经中国药科大学陆涛教授团队转让、后续自主研发的创新型小分子化学药物,主要用于白血病治疗。复星医药产业的控股子公司上海复星星泰医药科技有限公司2017年8月向CFDA提交了FN-1501于中国境内(不包括港澳台地区)用于治疗白血病的临床试验申请并获得注册审评受理。
复星医药此前公告中指出,目前中国境内尚无具有自主知识产权的、与FN-1501同靶点的药物上市,与FN-1501同靶点的新药在2017年首次于美国上市。推测FN-1501为小分子异柠檬酸脱氢酶2(IDH2)抑制剂。截至2017年9月,复星医药在FN-1501项目上的研发投入约2350万元。
关于IDH2
异柠檬酸脱氢酶(isocitrate dehydrogenase,IDH)是 一类在三羧酸循环中起重要作用的酶家族,通过抑制IDH2可以抑制多种促进细胞增殖的酶的活性。
2017年8月1日,FDA批准新基公司Idhifa (enasidenib)上市,用于治疗携带异柠檬酸脱氢酶2(IDH2)基因突变的成人复发或难治性急性髓系白血病(AML)。AML患者中携带IDH2突变的比例大约为8%~19%。
AML是一种快速进展的血液和骨髓肿瘤,发病率随年龄的增大而明显升高,中位发病年龄为66岁。在美国的AML中,适合接受骨髓移植的患者不足10%,而且大多数患者对化疗无响应并且会进展成复发或难治性AML,5年生存率大约20%~25%。美国2017年新确诊的AML患者大约有21380例,死亡病例大约10590例。
近日,黑龙江珍宝岛药业股份有限公司发布公告称,公司已收到国家食药监总局核准签发的通脉口服液《药品补充申请批件》。
批件的主要内容如下:
1、药品名称:通脉口服液
2、批件类别:药品补充申请批件
3、受理号:CYZT1600328
4、批件号:2017B03146
5、剂型:合剂
6、规格:每支装10ml
7、注册分类:中药
8、药品标准:WS3-B-3980-98-2015
9、原药品批准文号:国药准字Z20055499
10、原药品生产企业:哈尔滨天木药业股份有限公司
11、申请内容:药品生产技术转让(按照国食药监注[2013]38号文件情形三要求进
行药品生产技术转让)
12、药品生产企业:黑龙江珍宝岛药业股份有限公司
13、药品批准文号:国药准字Z20174074
14、药品批准文号有效期:至2022年12月20日
15、审批结论:根据《中华人民共和国药品管理法》及有关规定,经审查,本品此
次申请事项符合药品注册的有关要求,同意哈尔滨天木药业股份有限公司(生产地址 :
哈尔滨市利民开发区北京路1号)将通脉口服液药品生产技术转让至黑龙江珍宝岛药业
股份有限公司(生产地址:黑龙江省虎林市红星街72号),发给药品批准文号。
据了解,通脉口服液由丹参、川芎、葛根等中药组成。用于缺血性心脑血管疾病, 动脉硬化,脑血栓,脑缺血,冠心病,心绞痛。目前通脉产品有颗粒剂、合剂、片剂等剂型。目前,获得通脉颗粒生产批文的企业107家,获得通脉片生产批文的企业3家,获得通脉口服液生产批文的企业27家;通脉制剂2016年零售市场规模约9300万元,通脉口服液2016年市场规模约为120万元。
公司表示,该《药品补充申请批件》的取得,一方面丰富了公司心脑血管疾病药物的品类,另 一方面有利于提升公司在心脑血管疾病用药市场的竞争力,从而提高公司整体盈利水平。
作者 Ricky Shan
回首2017年,国内药企进军FDA硕果满满。以下,我们统计了2017年开始在美国进行I期临床的情况。本文仅包括2017年新开始的I期临床。直接进入II、III期的不在统计范围,已经获得IND批准未进入临床的也不在统计范围。若有遗漏,敬请指正~
1.丽珠医药——LZM009
此次在美国开展的研究是评估静脉注射LZM009用于晚期实体肿瘤患者的安全性和耐受性。
关于LZM009:
LZM009是注射用重组人源化抗PD-1单克隆抗体。LZM009是丽珠单抗具有自主知识产权的新药品种,该品种分子序列及用途专利已申请中国和PCT专利。
2. 益方生物(上海页岩)——D-0120
此次在美国开展的研究是一个随机、双盲、安慰剂对照、单剂量、剂量顺序递增用于评估D-0120在空腹的健康受试者上的安全性,耐受性,PK和PD。
3. 和记黄埔——Fruquintinib(HMPL-013)
此次在美国启动的研究是一项多中心、开放标签的1期临床试验,评估fruquintinib在美国晚期实体瘤患者中的安全性、耐受性和药代动力学。
关于Fruquintinib:
Fruquintinib(HMPL-013)是一款高度选择性血管内皮生长因子受体(VEGFR)1、2和3的口服抑制剂。与其它靶向治疗相比,通过口服剂量能24小时抑制VEGFR,且具有更低的脱靶毒性。其良好的耐受性,加上已被证明的药物相互作用安全性,使得它可以与其它癌症疗法进行合理组合,例如在正在进行的fruquininib的临床试验中,与化疗和其它靶向疗法组合。
4. 恒瑞医药——HTI-2088
此次在美国开展的研究是单中心,随机双盲,安慰剂对照,单次递增剂量(2.5mg, 3.75mg,5mg)试验。每一剂量将登记十名受试者。
5. 亚盛医药——APG-1252
此次在美国开展的临床I期剂量递增研究是用于确定最大耐受剂量(MTD),剂量限制性毒性(DLT),和临床II期的推荐剂量(RP2D)。相应的几个Ib/ II期研究也将在其后开展。
关于APG-1252:
APG-1252通过选择性抑制Bcl-2蛋白家族成员Bcl-2及Bcl-XL来恢复肿瘤细胞程序性死亡机制(细胞凋亡),从而杀死肿瘤。APG-1252是亚盛医药产品管线中在全球范围内第6个进入临床阶段的产品,也是企业第3个进入美国临床阶段的产品。APG-1252是首个在美进入临床、并由中国企业自主开发的的Bcl-2/Bcl-XL抑制剂。
6. 亚盛医药——APG-1387
此次在美国开展的临床I期剂量递增研究是用于确定最大耐受剂量(MTD),剂量限制性毒性(DLT),和临床II期的推荐剂量(RP2D)。APG-1387与pembrolizumab或联合化疗药物抗肿瘤作用的研究也将在其后开展。
关于APG-1387:
APG-1387是亚盛医药自主设计开发的、具有全球知识产权的新一代凋亡蛋白抑制因子(IAP)高效特异性抑制剂,是l类靶向小分子抗肿瘤药物,主要通过阻断IAPs的活性促进细胞凋亡的进程。目前国际上以IAP蛋白为靶点的药物均处于开发阶段,尚未有上市药物。APG-1387在中国和澳大利亚现均处于临床I期阶段。该品种为双聚体类化合物,可与IAP蛋白单体及双聚体均可形成稳定结合,有效克服了现有药物不能作用于IAP蛋白双聚体的缺陷。同时可实现临床一周仅给药一次的目标,是目前在研药物中开发前景最好的药物。
7. 百济神州——BGB-290
此次在美国开展的研究是评估BGB-290用于局部晚期或转移性实体肿瘤患者的安全性、药效及临床活动。
关于BGB-290:
BGB-290是一种针对PARP1及PARP2的高效选择性抑制剂。BGB-290正在作为单药及联合用药的疗法,用于开展针对多种癌症的研究,包括卵巢癌、前列腺癌、乳腺癌、多形性脑胶质瘤、小细胞肺癌及胃癌,其临床I期概念验证研究数据已在2015年AACR-NCI-EORTC会议上进行了报告。
8. 百济神州——BGB-290
此次在美国开展的研究是评估BGB-290联合放射治疗用于初诊或复发/难治性胶质母细胞瘤患者的安全性、药效及临床活动。
9. 百济神州——BGB-3111
此次在美国开展的研究是在健康受试者上确定rifampin(Part A)和itraconazole(Part B)对BGB-3111的药代动力学影响。
关于BGB-3111:
BGB-3111是百济神州开发的高选择性BTK抑制剂,之前公布的单药治疗多种晚期B细胞恶性肿瘤的I期研究显示,BGB-3111对慢性淋巴细胞白血病的总应答率高达93%,对套细胞淋巴瘤的应答率达到80%。百济神州曾在2016年第二季度财报中透露,BGB-3111获得了FDA的3项孤儿药资格认定。
10. 山东亨利——KBP-5074
此次在美国开展的研究是一个开放的,部分交叉,单剂量药代动力学(PK)用于评价剂量比例,以及kbp-5074片剂与胶囊制剂下在健康受试者的安全性/耐受性。从这项研究中获得的数据,加上临床前数据和药学数据,将为今后的研究提供剂量选择的基础。
关于KBP-5074:
KBP-5074是非甾体类盐皮质激素受体拮抗剂(MRA)。MRA类药物已被证明有独立的治疗慢性肾病、高血压和心衰的作用。慢性肾病最终导致肾功能衰竭,患者需要透析或器官移植。不仅造成患者的痛苦和高死亡率,而且对个人和社会造成巨大的经济负担。已上市和在研的MRA类药物因在治疗剂量易引发高血钾,导致心律失常甚至死亡,使其在慢性肾病患者中的使用受到极大限制,甚至禁用。全球有过亿患者目前无药可用,有巨大的临床未满足需求。
11. 绿叶制药——LY03005
此次在美国开展的研究采用交叉设计,在18和50岁之间的健康受试者空腹状态下分别单剂量口服80毫克LY03005片剂和50毫克Pristiq®片剂后,比较两者在受试者上的相对生物利用度。
关于LY03005:
LY03005是以缓释片形式制备的独家盐酸安舒法辛(一种5-羟色胺-去甲肾上腺素-多巴胺三重再摄取抑制剂(SNDRI))产品。与双重摄取抑剂相比,它起效更快,能有效改善快感缺乏、肥胖等副作用。
信息来源:clinicaltrials.gov; HPC整理编辑
------------------------------------------------------------
4、中药领域的一致性评价或将到来
医药行业仿制药质量和疗效一致性评价概念的提出,犹如医药行业投下一记重磅炸弹,一度引起制药企业,特别是中小企业的震惊、茫然、抵触和无所适从。
然而,仿制药一致性评价并不是我国独出心裁,而是欧美日等发达国家也曾经历十几年甚至二十几年走过的路程。我国开展仿制药质量和疗效一致性评价既有先例可循、可参考,又有在此基础上创新和发展。
一是开展一致性评价与规范医药行业生产经营活动相结合;二是开展一致性评价和创新药相结合;三是开展一致性评价和药品分类注册相结合。通过一致性评价能够更好地促进医药行业的创新和发展,规范和完善医药行业的用药安全和健全医药行业的监管体系。
针对我国批准上市的药品多达1.6万种,药品批准文号18.7万个,其中,化学药品批准文号12.1万个,大约97%是仿制药。全国不同规模的药品生产企业5000多家,低水平、重复生产和恶性竞争还普遍存在。
虽然我国仿制药发展取得了长足进步,也为有效解决了人民群众缺医少药做出了突出贡献,为维护公众健康发挥了重要的不可替代的作用。但是,这毕竟是历史,面对我国各行各业发展突飞猛进,中国特色社会主义进入了新时代,各界对药品质量和疗效的要求日益提高,我国仿制药存在检验合格,但是疗效不佳甚至无效等潜在问题也是越来越凸显,更有甚者影响到用药安全。仿制药质量和疗效一致性评价既不是打击制药行业、更不是颠覆制药行业,而是更好的促进制药行业整体提高和推动医药行业健康发展的必经之路。
一致性评价是国家战略,是系统工程。部分制药企业对一致性评价认识不足,还存在等待观望的态度,这是极为错误和危险的思维,往大了说是对人民健康不负责任,往小了说是不能与时俱进对企业和员工不负责任。
从2012年初至2017年底,历时六年时间,国家局以及各部门紧紧围绕一致性评价,花费大量精力、人力、物力先后起草下发各类征求意见、通知、公告、通告、方案、问答等文件72个,特别是近两年紧锣密鼓、频繁下发64个文件。这些都足以证明国家的高度重视和坚定的决心。
不容我们再等待、观望和消极懈怠,必须尽快行动起来,这既是国家对人民健康的期许,更是我们制药人的光荣责任和分内之事。作为制药企业自身要用延安整风的精神来对待仿制药质量和疗效一致性评价工作,要有严肃的态度、严明的纪律、严格的作风、科学的思维。
经过总局相关部门的大量认真细致工作,使制药企业对一致性评价工作从一头雾水、无所适从到有了比较详实的规范和遵循。围绕落实《国务院办公厅关于开展仿制药质量和疗效一致性评价的意见》(国办发【2016】8号)的安排,总局建立了一个方便备案的平台,成立了专家队伍,申报资料立卷审查、制定参比制剂的选择方案,确立了检验机构和临床试验机构以及“三改品种”,即改盐基、改剂型、改规格和豁免或简化人体生物等效性EB品种等政策和原则,并且都以文件形式固定下来,使之脉络逐渐清晰、流程相对严谨、环环相扣、具体操作可行。然而,如期完成一致性评价,笔者认为仍然有许多困难和不确定性。
一是按照总局要求的时间点,预测很难完成289个基药品种的一致性评价工作。根据总局办2016年3月31日发文再次强调:1、化学药品新注册分类实施前批准上市的仿制药,包括国产仿制药、进口仿制药和原研药品地产化品种,均须开展一致性评价。2、凡2007年10月1日前批准上市的列入国家基本药物目录(2012年版)中的化学药品仿制药口服固体制剂,原则上应在2018年底前完成一致性评价。
按照总局2012年11月22日《仿制药质量一致性评价工作方案》(征求意见稿)时间规划是:
2013年
建立参比制剂目录;构建口服固体制剂仿制药数据库,建立国家局、药品检验机构和企业之间信息传输系统;完成参比制剂的遴选与确认工作。
2015年
完成基本药物目录中固体口服制剂质量一致性评价的工作任务。也就是说在建立健全参比制剂目录的前提下,需要两年时间完成基药口服制剂的一致性评价工作。
然而,仿制药一致性评价办公室于2017年12月8日才发布《289目录品种参比制剂基本情况表》的通知,即便如此,还不够全面,描述的参比制剂898个规格,还有三改品种、研究中和因无厂家备案不推荐的。就算来年2月份全部参比制剂完成,按原设计时间计算可能也要到2020年2月份了,所以,时间依然很紧迫。
二是费用问题也是困扰企业的难题。目前,据初步了解,每个品种根据地域和品种差异,顺利完成每个品种等效性一致性评价,各种费用概算在500-1000万元区间。大家都很清楚基药品种除个别独家等品种外,都是属于低价格、低毛利品种,完成一个品种一致性评价,就该品种来讲基本上需要几年、甚至十几年收回一致性评价的投入成本,这也是为数不少的中小企业等待观望的主因,都期待着政府给与资金上的扶持或政策上的倾斜。
三是参比制剂企业自购依旧是个难题。据了解,不少中小企业对自购参比制剂心里没有底,有对政策的理解上的问题,有采购渠道上的问题等等,都希望国家局委托医药进出口公司或者医药行业协会统一采购,这样即便是费用高点但是心里踏实,也能节省时间和不必要的麻烦。毕竟总局有专家团队,对采购不到的或者已经下线的参比制剂,也有办法根据专家意见进行政策微调解决。
四是根据总局相关规定,在进行289基本用药以外的化学药品仿制口服固体制剂,自第一家通过一致性评价后,三年后不再受理其他药品生产企业的同品种一致性评价申请。也就说现在就可以进行289以外的仿制药一致性评价工作申请和立卷审查了,一旦某个品种通过一致性评价,三年内其它企业可以申请,三年后就等于文号注销了。
总而言之,一致性评价工作,时间紧,任务重,直接涉及企业利益甚至是生存,也涉及医药行业药品生产的深化改革步骤。可以断定2018年之后,随着一致性评价的顺利进行和不断深入展开,在不断总结经验和教训基础上,即将迎来对中药注射剂和生物制剂、中成药展开再评价工作,相当于中成药一致性评价,以此来完善和完成我国的特有的药品分类注册体系建设。
作者:中国医药兄弟联总会长、沈阳华卫集团执行总裁 王振林
---------------------------------------------------------------
5、全景:国内wAMD相关眼科药物在研一览
作者 Daniel
wAMD,湿性年龄相关性黄斑变性。通俗来讲:湿性年龄相关性黄斑变性会长异常的新生血管,是因为新生血管因子,也就是血管渗漏因子的升高,视网膜才会长异常的新生血管。给患者打新生血管抑制剂,也就是抗VEGF抑制剂,可以快速的抑制新生血管,减轻视网膜组织的水肿和渗出,也可以快速的提高视力。目前新生血管抑制剂,是大多数眼科医生治疗湿性年龄相关性黄斑变性首选的药物。目前国内主流药物为诺华的雷珠单抗和康弘的康柏西普。拜耳和再生元的阿柏西普预计2018年国内获批。
下文仅为笔者平时搜集记录,欢迎各位补充。
1、目前在做临床的药物
2、目前在申报临床的相关眼科药物
3、成都康弘开展的相关临床
4、烟台荣昌生物
(1)相关专利,血管新生因子融合蛋白在制备用于治疗与血管新生相关的眼部疾病药物中的应用:CN105983093A。荣昌专利本发明涉及多种血管新生因子融合蛋白在制备用于治疗与血管新生相关的眼部疾病药物中的应用,如糖尿病视网膜病变,年龄相关黄斑变性等。更具体地,本发明涉及VEGF受体和FGF受体的融合蛋白在制备用于治疗与血管新生相关的眼部疾病药物中的应用。本品是一种VEGF受体和FGF受体与人免疫球蛋白Fc段基因重组的融合蛋白,属于受体抗体融合蛋白。2013年9月列入国家“重大新药创制”科技重大专项“十二五”课题。
本发明人出人意料地发现拮抗血管新生因子(特别是拮抗FGF和/或双重拮抗VEGF和FGF)的大分子蛋白药物能够更好地抑制血管的新生,在临床上得到更好的治疗效果。本发明人通过构建多种包含不同的VEGFR片段、FGFR片段和IgGlFc的融合蛋白,获得了对VEGF和FGF(尤其是FGF-2)具有高度亲和力的融合蛋白以及对FGF(尤其是FGF-2)具有高度亲和力的融合蛋白。而且,本发明人证明本发明的融合蛋白在用于治疗血管新生相关眼部疾病时的治疗效果显著优于现有技术中针对VEGF单靶标的药物(例如美国Regeneron公司研发的VEGF-Trap)。特别地,本发明的融合蛋白在糖尿病视网膜病(例如STZ诱导的DR大鼠模型)、年龄相关的黄斑变性(例如激光致恒河猴脉络膜新生血管抑制模型)、早产儿视网膜病(例如高氧诱导的视网膜病OIR小鼠模型)中表现出出人意料地显著优于VEGF单靶标药物的治疗作用。
(2)桥连的双特异性融合蛋白,CN106084062A,本发明涉及桥连的双特异性融合蛋白,具体地,本发明涉及改良的拮抗血管新生诱导因子的融合蛋白及其用途,更具体地,本发明涉及VEGF受体和FGF受体的融合蛋白及其在治疗与血管新生相关的疾病中的用途。
5、雷珠单抗生物类似药
(1)国内目前有齐鲁制药申报雷珠单抗(Lucentis,Ranibizumab)生物类似药,重组抗VEGF人源化单克隆抗体Fab注射液,2016年9月拿到临床批件。另外齐鲁还有重组人血管内皮生长因子受体-抗体融合蛋白注射液在研。
(2)百奥泰生物拥有雷珠单抗生物类似药。
已将本品与原研药进行系统的比较研究,各项质量研究结果均表明该产品的质量属性与原研药雷珠单抗高度相似。本产品目前已筛选出候选抗体,产量达到国际水平。拟适应症:糖尿病性黄斑水肿(DME)、视网膜静脉阻塞继发黄斑水肿(RVO-ME)、湿性年龄相关性黄斑变性(wet-AMD)、糖尿病性视网膜病变(DR)、病理性近视继发脉络膜新生血管(mCNV)。相关专利:一种高稳定的治疗VEGF相关疾病的人源化抗体制剂,CN105079804A。
(3)此前华东医药在2016年报中表示,公司同样在开发雷珠单抗,处于临床前阶段。
6、复宏汉霖抗VEGF单抗新增适应症获受理
2017年12月,复宏汉霖自主研发的HLX04——重组抗VEGF人源化单克隆抗体注射液用于治疗湿性年龄相关性黄斑变性和糖尿病性视网膜病变适应症获CFDA临床试验注册审评受理。
据悉,HLX04 - 重组抗VEGF人源化单克隆抗体注射液是贝伐珠单抗(Bevacizumab,商品名:安维汀®)的生物类似药,可通过抑制VEGF的活性来控制新生血管的形成,进而控制相关的疾病。2015年12月,HLX04获得结直肠癌适应症的临床试验批准;随后,HLX04新增非小细胞肺癌的适应症,并于2016年5月获批临床。截至目前,HLX04的结直肠癌适应症和非小细胞肺癌适应症已经处于一期临床试验阶段。
7、东方百泰
2017年5月,自主研发的单抗药物“重组人源化抗VEGF单克隆抗体注射液”获得CFDA颁发的临床批件。获批的临床适应症为:湿性(新生血管性)年龄相关性黄斑变性(AMD)。重组人源化抗VEGF单克隆抗体,系通过基因工程技术在哺乳动物细胞(CHO)表达系统生产,用于治疗湿性(新生血管性)年龄相关性黄斑变性(AMD)。本品与VEGF有较高的亲和力,抑制VEGF与其受体VEGFR1和VEGFR2的结合。VEGF与其受体结合,可导致血管内皮细胞增殖和新生血管形成,以及增加血管渗透,这些被认为与新生血管性年龄相关性黄斑变性(AMD)的进展相关。
8、三生制药
三生国健,2017年10月,由集团开发的重组人源化抗血管内皮细胞生长因子(VEGF)单克隆抗体注射液,获CFDA出具的新药临床试验申请批件。三生制药公告表示:拟开发该产品用于治疗新生血管性年龄相关性视网膜黄斑变性(AMD)。AMD是老年人不可逆性失明的主要病因之一。美国的一项流行病学研究显示:在52-64岁人群中,AMD的发病率约为2%。而75岁或以上人群中的发病率则上升至约为28%。近年来随着中国老龄化人口的增加,AMD在中国的发病率大幅上升。
9、苏州思坦维
hPV19单抗,公司此前表示:临床前研究显示,hPV19单抗与VEGF抗原的结合力及生物活性显著强于市售同类进口药物雷珠单抗(Ranibizumab)。hPV19单抗眼用注射液项目于2014年获江苏省科技厅立项(立项编号:BE2014635),获江苏省政府科技基金及苏州工业园区政府科技基金支持。
10、治疗wAMD的小分子
目前国内笔者搜索到的小分子治疗wAMD的只有:CM082。截止目前,卡南吉(贝达药业)已经获得包括年龄相关性黄斑变性在内的共4个眼科适应症的临床批件,还包括糖尿病并发黄斑水肿/视网膜静脉阻塞继发黄斑水肿/病理性近视脉络膜新生血管。
此前,Tyrogenex在国际知名医学期刊——JAMA(美国医学会杂志)发表题为《Oral Tyrosine Kinase Inhibitor for Neovascular Age-Related Macular Degeneration: A Phase 1 Dose-Escalation Study》(中文标题为《口服酪氨酸激酶抑制剂用于新生血管性年龄相关性黄斑变性:一项I期剂量爬坡研究》)的文章,披露了X-82治疗新生血管性年龄相关性黄斑变性(AMD)的I期临床研究结果。
11、其他专利:
常州亚当生物:抗VEGF单克隆抗体Fab片段Vasculizumab及其应用,CN102286101B:将识别VEGF的抗体片段在大肠杆菌中表达,其抑制眼科血管新生的效果与全抗分子相比无明显差异,而该Fab片段分子量低,容易到达靶部位;Fab片段可以在大肠杆菌中大规模生产,效率高且成本相对较低 。
一种抗VEGF/PDGFRbeta双特异性抗体及其应用,CN102250249A。
一种抗VEGF抗体及其应用,CN103739710B,中国人民解放军军事医学科学院基础医学研究所。
靶向VEGF双特异性抗体及其用途,CN105481981A,中国人民解放军第二军医大学:本发明在Bevacizumab及全人源单克隆抗体B5E3基础上构建一个针对VEGF两个表位的双特异性抗体Bev-B5E3BsAb,实验表明其不与Bevacizumab竞争结合VEGF,推测其表位是不同于Bevacizumab的新表位。同时,与其两亲本单克隆抗体相比,能够更为有效地结合VEGF。此外,体外实验中显现出更强地抑制人脐静脉血管内皮细胞(HUVEC)增殖的能力;动物实验表明其能够显著抑制肿瘤的生长。因此,该双特异抗体可望发展成为用于肿瘤以及眼科疾病治疗或诊断新型抗体制剂。
------------------------------------------------| 欢迎光临 药群论坛 (http://yaoqun.net/) | Powered by Discuz! X3.2 |