表1. FDA从2003到2011年要求实施“有因”检查的原因分类
原因分类 | 要求检查数 | 百分比, % | |
1 | 数据可靠性与有效性问题 | 55 | 60.4 |
2 | 再分析的样本量过多 | 7 | 7.7 |
3 | 被检查单位此前有不良检查记录 | 11 | 12.1 |
4 | 文件不完整 | 9 | 9.9 |
5 | 研究设计与实施不当 | 4 | 4.4 |
6 | “其他” | 5 | 5.5 |
合计 | 91 | 100 | |
表2. 数据可靠性与有效性问题方面的具体原因
具体原因 | 要求检查数 | 百分比,% | |
1.1 | 要求实施检查以确保数据的准确性 | 34 | 61.8 |
1.2 | 申办者递交的信息与FDA获得的其他信息存在分歧 | 2 | 3.6 |
1.3 | 研究方案偏离 | 5 | 9.1 |
1.4 | 方法验证不充分 | 8 | 14.5 |
1.5 | 递交的信息不一致/相互矛盾 | 6 | 10.9 |
合计 | 55 | 100 | |
3.结果与讨论
检索到2003年1月到2011年12月 FDA 对ANDA实施了累计90项“有因”检查。这些检查涉及66个ANDA申请。在此期间,FDA共受理了7,207个ANDA申请。因此,2003年到2011年期间,被要求实施“有因”检查的比例占所有ANDA申请的0.9%。需要说明的是,除“有因”检查外,FDA还会实施“常规”检查,该检查可在没有发现行为不端的情况下启动或者作为投诉的跟踪。通常,“常规”检查用于对支持注册申请,如新药上市申请、ANDA及新药临床申请等开展研究的评估。对“常规”检查的评估不属于本文所探讨的范畴。
在通常情况下,一项典型的药代动力学生物等效性研究的参与者包含受试者服药和血样采集的临床研究单位和对血样进行分析以测定药物浓度的分析单位。
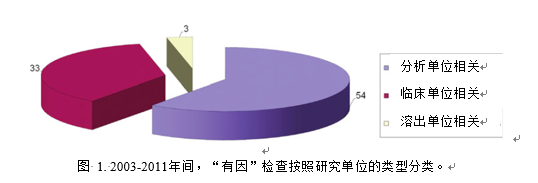
同时临床终点生物等效性研究可能会涉及多家临床研究单位。对于某些剂型,如片剂或胶囊,ANDA申办者也会进行比较溶出试验为生物等效性提供支持。在90项“有因”检查中,有54项涉及分析单位的检查(图1)。此类检查主要是针对所涉及的生物分析方法的准确性与可靠性。另有33项涉及临床研究单位的检查。此类检查旨在确定临床研究的开展是否合规,受试者的权利、安全及权益是否得到了保护。有3项检查涉及溶出研究单位。要求对溶出单位实施检查的原因是FDA审评员发现ANDA申办者递交的溶出结果与监管部门从其他申请资料掌握的参比制剂溶出的信息存在差异,甚至ANDA申办者的溶出试验报告本身就存在不一致。
我们随后对启动“有因”检查的常见原因进行了分析。通过研究发现,FDA要求实施“有因”检查最常见的原因与“数据可靠性和有效性问题”相关,占要求实施检查总数的60.4%(表1)。位列第二的要求实施“有因”检查的最常见原因是被检查单位此前有不良检查记录。这种情况下,FDA审评员要求检查的目的是为了核实以往对该单位检查时发现的数据可靠性类似不良问题在正在审评的研究中仍然存在。该原因占要求实施检查总数的12.1%。“文件不完整”也属于严重的问题,占检查总数的9.9%。例如,如果申办者没有保留研究受试者充分且准确的病历,并且FDA审评员确认此类数据不完整可能影响研究的结果,审评员可能要求实施检查以便澄清及确认。除上述最常见的三种原因外,我们对其他导致“有因”检查的原因也进行了分析。此类原因与申请材料中发现的研究设计、研究实施、标准规程、数据报告及样本再分析缺陷有关(见表1)。对于无法明确归类的原因,我们将其列为“其他”类。属于此类型的“有因”检查包括一例受试者在空腹生物等效性研究中死亡的病例。FDA审评员对申办者的研究方案及医学记录进行检查,并未发现有严重疏忽或医疗管理不善的证据。但是审评员对于死亡事件及严重不良事件仍表示担忧。因此,为了进一步核实临床试验和医学记录、研究者的资质以及所用标准操作规程(SOPs)对评价和管理受试者安全问题和不良事件的适用性,审评员要求对其实施“有因”检查。
表3. FDA要求实施“有因”检查的原因举例
“有因”检查的原因 | 实例 | |
1 | 数据可靠性和有效性问题 | 见表4 |
2 | 再分析的样本量过多 | 在空腹及进食研究中,大量样本分析批被中断和/或分析物被重分析;申办者对中断及重分析没有提供充分的依据。此外,还有大量的样本中分析物重新积分,但是申办者没有对重新积分提供充分的依据。 |
3 | 被检查单位此前有不良检查记录 | 此前在对该研究单位进行检查时发现许多受试者研究样本都存在完整性的问题。 |
4 | 文件不完整 | 申办者在研究受试者的进度记录本上没有保留充分且准确的病案历史。 |
5 | 研究设计与实施不当 | 再次给药研究中对照受试者样本量不足,在实施异常值检测时缺乏可用的标准操作规程。此外,分析方面的缺陷还包括了质控浓度选择不当。 |
6 | “其他” | 要求对临床机构进行检查是基于已知特定药物引起严重不良事件以及研究受试者住院。 |
表4. 因“数据可靠性和有效性问题”而要求实施“有因”检查举例
“有因”检查的原因 | 实例 | |
1.1 | 要求实施检查以确保数据准确性 | 要求实施检查以核实受试者X在生物等效性研究中所有采样时间点所测得的药物浓度都低于定量下限;核实公司规程在研究单位是否得到严格执行以确保受试者用药剂量,并且确认不存在其他可导致生物等效性研究结果无效的分析缺陷。 |
1.2 | 申办者与FDA内部数据存在分歧 | 分析物的AUC0-t、AUC∞和Cmax等参数值与FDA从其他内部获得的数据存在较大的偏离。 |
1.3 | 研究方案偏离 | 申办者没有确保研究按照研究计划执行。 |
1.4 | 方法验证不充分 | 对于所关注的分析物缺少交叉验证的研究数据 |
1.5 | 提交资料中信息不一致/相互矛盾 | 申办者提供的研究日期。自相矛盾,并对受试者样本的储存时间和稳定性产生影响。 |
我们进一步对“数据可靠性和有效性”方面的原因进行了具体的分析。在此类别中,最常见的原因是为了核实申办者递交的生物等效性研究的数据是否准确。例如,生物等效性审评员注意到某例受试者在生物等效性研究中所有采样时间点所测得的药物浓度都低于定量下限。随即要求对其实施检查,以核实临床单位是否严格执行了公司的规程以确保受试者的用药剂量,并且核实不存在其他可导致生物等效性研究结果无效的分析缺陷。“方法验证不充分”是此类型中第二大常见问题。此外,如果FDA审评员发现申办者递交的材料中有信息不一致或相互矛盾且无合理解释的,则会要求对其实施检查。除上述提到的原因外,还有小部分检查则是基于潜在的不合规问题和不足,例如:研究方案偏离,研究者提供的信息与FDA获得的其他信息存在无法解释的不一致(见表2)。
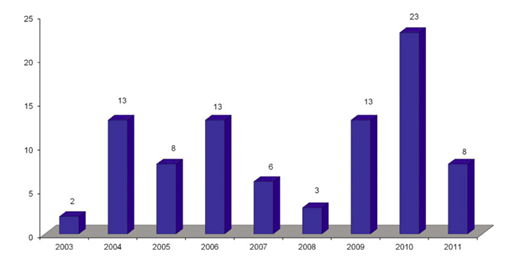
图2. 2003年到2011年每年要求“有因”检查的数量
需要说明的是,本次研究中提及的多项检查并非是因为单一的问题而要求实施检查的。在通常情况下,由于研究报告中出现的多种问题而暴露的系统性问题,很可能会引起FDA审评员对研究是否有效的担忧,从而启动“有因”检查。例如,FDA审评员发现相同样本因为不同的原因多次重复检测,并且申办者对此类重测未提供充分的依据。此外,FDA审评员在审阅分析报告细节时,发现申办者提供的分析研究存在明显的不一致、不相关以及无效的问题。综合上述所有原因,审评员做出了要求“有因”检查的决定。表III和表IV呈列了FDA审评员要求实施“有因”检查的原因的某些典型案例。
对于每年ANDA申请中要求实施“有因”检查的数量,并没有明显的趋势。2003年,仅要求2项“有因”检查,但在2010年,则实施了23项“有因”检查(图2)。在9年研究期间,平均每年要求10项“有因”检查。
检查结果分为以下三种:无需采取措施(NAI),自愿采取措施(VAI)以及采取官方措施(OAI)。NAI是指在检查时没有发现违规情况或操作,或者违规情况并不需要采取进一步的监管措施。VAI是指在检查中发现有偏离法规的情况或操作,但是申请人随即采取自愿措施对此类情况或操作进行了整改。OAI则是指检查时发现违规情况/操作,建议采取监管和/或管理方面的措施。如图3所示,在本研究所考察的“有因”检查中,有15项属于NAI,有53项属于VAI,而15项则属于OAI。图3还显示,有7项尚未得出结论的检查。从这些数据可以看出,FDA审评员要求实施的“有因”检查中,有75%的检查会被合规办公室定性为存在潜在问题(VAI)或严重问题(OAI)。此类生物等效性研究将要求申办者采取相应的官方或自愿的纠正措施。

图 3. 从2003年到2011年“有因”检查结果的类型
4.结论
本项研究探讨了FDA 对参与ANDA生物等效性研究的临床研究单位、分析研究单位及溶出研究单位实施“有因”检查的常见原因。导致采取官方及自愿纠正措施结果的检查可能会延长申办者的申请获得批准的时间,因此应当予以避免。希望通过此文章的发表可以为申请人在力争符合FDA法规方面提供方法,从而能够促进安全有效的药物更快地获得批准。
(来源:Li, B.V., Davit, B.M., Lee, C.H. et al. AAPS J , 2013 Jan, 15(1): 10-4,作者:美国CDER仿制药办公室李冰等,翻译:王安娜,审校:董江萍)
原文刊登于《国际药品检查动态研究》第2卷第1期(总第4期),2017,P20